Pustular psoriasis study protocol
1. Background and Rationale
The term pustular psoriasis refers to a group of severe and disabling, neutrophilic dermatoses with major physical, psychological and social morbidity. PP may be generalised and potentially life threatening (generalised pustular psoriasis (GPP)) or localised, when it is characterised by chronic, painful, pustulation on the palms and soles (Palmar-Plantar Pustulosis, PPP) or fingers (Acrodermatitis Continua of Hallopeau, ACH). Pathogenic alleles have been identified in the IL36RN gene, but mutations at this locus only account for a fraction of disease cases. Besides, the limited size of patient datasets has so far hindered the execution of studies correlating IL36RN genotype with clinical phenotype. Thus, there is a clear need for the recruitment of extended patient resources, which will facilitate the discovery of further disease genes, the study of genotype-phenotype correlations and the identification of biomarkers of drug response.
2. Trial Objectives and Design
The aim of this study is to address the issues outlined above, by pursuing the following objectives:
2.1 Objective
To secure a comprehensive collection of biological samples (DNA for all subjects, RNA, cells, on designated subsets) to match corresponding clinical datasets, thereby establishing a critical resource for future use by investigators, to improve outcomes in pustular psoriasis
- To contribute to the identification of novel genetic determinants for pustular psoriasis
- To help determine how PP genes interact with each other and with environmental disease triggers
- To help determine how PP genes influence the presentation of the disease and the response to systemic treatment
2.2 Study Design
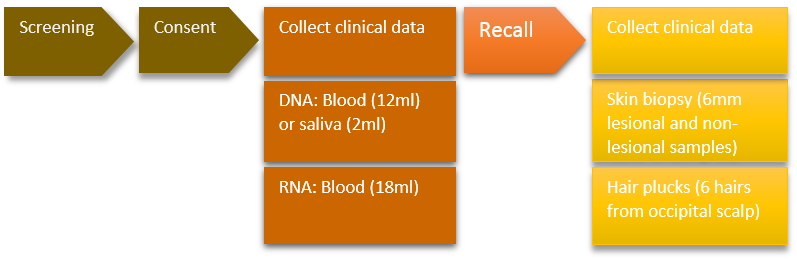
There are 4 main components ('phases') to the study design;
1.1 Case and control ascertainment
Patients with pustular psoriasis will be recruited from dermatology clinics and using information from clinical notes and databases. Healthy volunteers will be recruited via advertisements on the King's College London (KCL) website.
Following written, informed consent, a blood or saliva sample (see section 4.2B) will be collected for DNA extraction. Phenotype information will also be recorded on the 'psoriasis case report form' (see attached).
1.2 Analysis of DNA
Exome sequencing and candidate gene analysis will be performed on DNA to identify genetic variants in pustular psoriasis-associated genes. All participants are informed, both verbally (prior to seeking consent for the participation in the study) and in the patient/participant information sheets that their DNA is analysed solely for the purposes of psoriasis research and the analysis will not provide any information on their risk of developing other diseases.
2.1 Recall of cases
Psoriasis patients recruited in phase 1 who are found to have variants in psoriasis-associated genes will be contacted (see section 3.3E) and asked if they are willing to be invited back to clinic to provide further biological samples, for the purposes of functional assays.
2.2 Recall of healthy volunteers
Healthy volunteers without variants in psoriasis-associated genes who were recruited in phase 1 will be contacted and asked if they are willing to be invited back to clinic to provide further biological samples.
2.3 Recall of relatives of patients
If a patient reports a family history of psoriasis or has a detectable variant in a psoriasis-associated gene, they will be asked to deliver study invitations and participant information sheets to relevant family members. Relatives who wish to participate will then be contacted by a member of the study team and invited to clinic. Following written, informed consent, a blood or saliva sample will be collected for DNA extraction. Phenotype information will also be recorded on the 'psoriasis case report form'.
All subjects who have been recalled (phase 2) will be asked if they are willing to provide skin biopsies (or hair plucks if skin biopsy is not feasible) in addition to blood. Samples obtained from cases and controls will be compared in order to determine the biological impact of psoriasis-associated variants.
The genetic variants and functional effects will be correlated with phenotype and drug response.
3. Subjects
3.1 Inclusion Criteria
- Individuals able to give written informed consent
- Aged ³16 years
- Patients with psoriasis (as diagnosed by a dermatologist) or
- Healthy volunteers
3.2 Exclusion Criteria
- Individuals unable to give written informed consent
- Individuals who have received a blood transfusion within 4 weeks (where DNA is being secured via whole blood).
3.3 Recruitment strategies and procedures
Eligible patients will be identified via the clinical database, clinic lists or clinical notes. The patients may be approached via post prior to their clinical visit using the 'Patient Invitation Letter' to allow the patient extra time to consider the study. The letter will include contact details for the chief investigator. The 'Patient Information Sheet' will be included with the letter.
Eligible patients attending a dermatology clinic will be approached by a member of the hospital research team who will discuss the study with them. They will be given a ‘Patient Information Sheet’ and given sufficient time to decide if they wish to take part in the research. If they wish to participate, a member of the research team will obtain written informed consent from the patient, which includes permission to access patient notes for phenotype information and for recall. Patients will be asked to provide the relevant biological samples, together with clinical data as dictated by the protocol.
Control subjects will be recruited through advertisements on the KCL website and provided with study invitations and participant information sheets. If they wish to participate, a member of our study team will contact the subject (by telephone/letter/email) and invite them to clinic to provide the relevant biological samples, together with clinical data, as dictated by the protocol. They will also be consented for recall.
If a patient reports a family history of psoriasis or has a detectable variant in a psoriasis-associated gene, they will be asked to deliver study invitations and participant information sheets to relevant family members. If they complete the reply slip on the participant invitation letter they will then be contacted by a member of the study team and invited to clinic to provide biological samples and clinical data, as dictated by the protocol. They will also be consented for recall.
If a specific genetic variant is detected in psoriasis patients or family members, we may wish to recall them for further clinical phenotyping and collection of biological samples for functional studies (skin biopsy, or hair plucks if skin biopsy is not feasible). We will also recall healthy volunteers without variants in psoriasis-associated genes as controls. It is likely that a subject with the relevant genetic variant will only be recalled once. This recall is entirely voluntary as stated in the Patient/Participant Information Sheet. The subject will sign a separate section on the Consent Form indicating their agreement. If it is necessary to recall the subject, a member of the study team will contact them by letter, email or telephone or approach them at their next clinic visit and ask if they are still willing to provide further clinical data and biological samples.
3.4 Withdrawal of subjects
A participant may voluntarily discontinue participation in this study at any time. This will not affect his/her current or future treatment. If the participant wishes to discontinue participation, the 'withdrawal of consent form' will be completed and kept in the study file. The chief investigator or co-investigators may also, at his or her discretion, discontinue the subject from participating in this study at any time. If a biological sample has been collected and it is determined that the subject does not meet the inclusion criteria for participation, or if the patient withdraws consent from the study, then the study physician must complete the appropriate documentation to request sample destruction (see 'request for sample destruction' document). It is the responsibility of the chief investigator to destroy the sample and to keep a record of that destruction in the study file.
4. Investigational plan
4.1 Visit schedule
A. Patients with psoriasis, relevant family members, healthy volunteers
4.2 Measurements And Evaluations
The following phenotype data will be collected and recorded on the 'psoriasis case report form:
- Demographics
- Pustular Psoriasis subtype
- Presence of psoriatic arthritis and other co-morbidities
- Past history of psoriasis (age of onset, clinical course, details of disease flares including possible triggers and markers of systemic inflammation)
- Date, dose, effectiveness and reason for discontinuation of previous therapies
- Ongoing concomitant therapy
- Family history of psoriasis (details of family members, age of onset, treatment, parental relatedness)
- Smoking status and alcohol intake
- Clinical assessments: weight, waist, height, blood pressure and disease severity (psoriasis area severity index (PASI) if plaque psoriasis or palmoplantar pustulosis PASI (ppPASI) if pustular variant), physician's global assessment (PGA), body surface area (BSA), dermatology life quality index (DLQI)
1. Blood samples – collected from all participants
- After receiving the participant's written informed consent, 12mls of blood will be collected in pink top EDTA vacuette tubes for DNA extraction. The blood is collected in EDTA coated tubes as this prevents blood from clotting. It is preferable to using heparin as an anticoagulant, as heparin may interfere with subsequent amplification of DNA by PCR.
- Participants will also be asked to provide 18ml of blood for RNA extraction, which will be used for (functional) expression studies in single immune cell types. This will be collected in purple top EDTA vacuette tubes.
- Blood will be transported to the Division of Genetics and Molecular Medicine (King's College London), Guys Hospital, London by courier or by post where it will be stored. Storage of biological material will be in accordance with section 4.4; transport of samples will be in accordance with SOPs.
2. Saliva samples - obtained from all participants for whom blood collections are not feasible
- Where blood collection for DNA is not feasible, saliva samples may be collected. A total of 2mls of saliva will be collected using the Oragene®-DNA self-collection kit from DNA Genotek (www.dnagenotek.com). Saliva DNA is stable in this form at ambient temperature.
- Saliva will be stored within the Division of Genetics and Molecular Medicine (King's College London), Guy's Hospital, London.
- In exceptional circumstances where a subject (for example psoriasis patient identified from the clinical database or family member) is not able to attend clinic, they may be asked to provide a saliva sample for DNA via post. The chief investigator will approach them by post, including the ‘Patient Information Sheet’ and a reply slip which they will be asked to return if they wish to participate. If they reply agreeing to participate they will then be sent a consent form and a DNA sample donation pack (for saliva) and asked to sign and return the consent form and return the sample pack to Division of Genetics and Molecular Medicine (King's College London), Guy's Hospital, London via Royal Mail Freepost service. Those subjects who do not reply to the initial letter will be followed up four weeks later by a member of the study team, by telephone, email or letter, and asked if they wish to participate.
3. Skin biopsy and/or hair plucks - collected from subjects with genetic variants in psoriasis-associated genes and control subjects without variants in disease-associated genes
- With the participant's written, informed consent, a skin biopsy will be performed.
- Two 6mm punch skin biopsies will be obtained from a lesional site and a non-lesional site using standard protocols for biopsy sampling. Lesional biopsies will only be performed in subjects with psoriasis. Primary sites for biopsies are the lower back and buttocks. If no lesions are present at these sites, biopsies may be taken at other sites. The site of biopsies will be recorded.
- If a skin biopsy is not feasible, 6 hair plucks will be performed using tweezers to gently pull the hair out. Ideally the occipital part of the head should be sampled.
Skin biopsies and hair plucks will be stored in accordance with section 4.4.
4.3 Laboratory studies
DNA from pustular psoriasis patients, healthy individuals and family members will be extracted from blood samples using standard methodologies. If participants are unable to provide blood, DNA will be extracted from saliva samples. Using exome sequencing and Sanger sequencing (for candidate genes), novel psoriasis-associated genetic variants will be identified.
On the basis of the genetic variant detected, participants (psoriasis patients, family members, subjects with loss of function variants and controls) will be invited to clinic to obtain biological samples for functional analyses. Through functional assays that determine the consequences of disease-associated variants on cell function, we aim to gain insight into pathogenic mechanisms. This work will utilise tissue harvested from skin biopsies and/or hair plucks and RNA extracted from blood. Primary cell cultures and immortalised cell lines may be established for functional assays. It is specified in the patient/participant information sheet that the participant will not have any financial benefits or rights over these cell lines.
The benefits of this research include diagnostic, prognostic and therapeutic advances in psoriasis, following the identification of underlying causal genetic variants, and a better understanding of their functional consequence.
4.4 Storage of Biological Specimens
Blood, saliva, skin and hair plucks will be stored in a secure facility in the Division of Genetics and Molecular Medicine, King’s College London, 9th Floor, Guy’s Tower, Guy’s Hospital, London, SE1 9RT.
Whole blood for DNA will be stored at -20°C until extraction. Saliva will be stored at ambient temperature until DNA extraction. Whole blood for RNA, skin samples and hair plucks will be processed immediately after collection. DNA will be stored long term at -20°C and RNA at -80°C.
4.5 Sample size
For the discovery of novel genetic variants in pustular psoriasis-associated genes by exome sequencing (see section 2.2, phase 1), power calculations indicate that a patient sample size of 122 will provide 80% power to detect a disease-associated genetic variant with a minor allele frequency of 0.01 and odds ratio of 5 (threshold p value is 1x10-6). We therefore plan to recruit at least 150 psoriasis patients.
To investigate the functional consequence of disease-associated genetic variants (see section 2.2, phase 2 and 3), T test power calculations indicate that 16 psoriasis cases and controls (for each variant investigated) will provide 80% power to detect a 25% difference in functional effect given variability of 25% in results (threshold p value 0.05). We therefore plan to recruit at least 20 psoriasis cases, 20 controls for the functional characterisation of each disease associated variant.
5. Assessment of Safety
5.1 Possible adverse effects
The possible adverse effects for the subjects are related to the procedures. When taking blood samples discomfort, hematoma and infection may occur. Skin biopsy carries the risk of bleeding, discomfort, infection and scarring. After injection of local anaesthesia, intolerance reactions might also very rarely occur. Based on our long lasting experience with the described routine procedures we consider the risks of all adverse effects as low.
5.2 Safety Reporting
This non-CTIMP study does not impact patient treatment and has low risk of causing adverse events. Where an adverse event occurs due to or during the collection data for the study, such as infection or injury caused by blood extraction or syncope, the chief investigator will report to the main Research Ethics Committee any SAEs in line with the National Research Ethics Service standard operating procedure on reporting of SAEs as defined in the 'standard operating procedure for safety reporting for non-CTIMP studies'.
6. Administrative Aspects
6.1 Good Clinical Practice
The planning, conduct and reporting of this study will be in accordance with Good Clinical Practice (GCP).
6.2 Declaration of Helsinki and Ethical Review
The study will be performed in accordance with the principles stated in the Declaration of Helsinki, 2008.
The final study protocol, including the informed consent form, will be approved by an Ethics Committee before enrolment of any subjects into the study. The opinion of the Ethics Committee will be dated and given in writing. A list of those present at the committee meeting (names and positions) should be attached whenever possible. All correspondence with the Ethics Committee will be filed in the Investigator Site File
6.3 Subject Information and Consent
The chief investigator will ensure that the subject is given full and adequate verbal and written information about the nature, purpose, possible risk and benefit of the study. If the participant does not speak English, a translator who is fluent in both English and the participant's spoken language will be provided.
The subject will be given an appropriate amount of time to consider their participation in the study and the opportunity to ask questions. Subjects will also be notified that they are free to discontinue their participation in the study at any time.
Written informed consent will be obtained from all subjects before enrolment into the study. The subject should retain a copy of the Participant/Patient Information Sheet and the signed informed consent form. A letter will also be sent to the patient's GP, with the patient's consent, informing them that their patient has participated in this study.
6.4 Subject Protection
Only members of the clinical care team will have access to patient records and make the initial approach to patients, if they meet the inclusion criteria. Subjects will be assigned a linked-anonymised study number to ensure subject confidentiality throughout the duration of the study. Only the subject number will be referenced on study related documents, samples, study publications and presentations. All samples will also be labelled with a unique ID in an anonymised form. The study team members outside the clinical team will have access only to anonymised data.
The chief investigator will be responsible for keeping a Subject Identification Log of all subjects enrolled into the study, their corresponding study number and sample IDs. This information will be kept on a secure server in a password protected file and will only be available to the chief investigator and the study personnel who are directly obtaining clinical data.
The subjects will also be informed in writing that authorised representatives of the Regulatory Authorities may require access to those parts of the hospital/practice records relevant to the study, including medical history, for verification of data.
6.5 Direct Access to Source Data and Documents
Key investigators/collaborators of the project will be the only personnel with access to patient and study data and all data will be stored in accordance with the Data Protection Act, 1998. The chief investigator will have overall control of, and act as the custodian for all data for the full duration of the study. The data will be available for internal monitoring (verification of data using hospital notes against the information recorded in the psoriasis case report form).
6.6 Data management and storage
All data generated will be backed up regularly in local storage facilities with scheduled back up plans according to local institutional policy.
6.7 Participant reimbursement
Control subjects who participate in this study will be reimbursed the cost of their travel (if applicable) and £30.
7. Other Study Issues
7.1 Monitoring
Monitoring includes the verification of data using source data (hospital notes) against the information recorded in the psoriasis case report form. By participating in this study the investigators agree to comply with guidelines for GCP. The chief investigator will be responsible, in accordance with local research governance procedures, for maintaining the site investigator file, ensuring study data is recorded in the source notes for each patient and for the monitoring of clinical data to ensure accurate data capture.
7.2 Training
The chief investigator will ensure that appropriate training relevant to the study, including GCP, is given to the medical, nursing and research staff involved, and that any information of relevance to the performance of this study is forwarded to the co-investigators and other staff involved.
7.3 Study Timetable
Estimated start date (first patient in) – January 2015
Estimated end date (last patient out) – January 2020
7.4 Subject Medical Records
For every subject taking part in the study, source documents should clearly indicate at least:
- that the subject participated in the study, e.g. by including subject identification (subject number) and study identification (study code or other)
- diagnosis(es) (past and current; both the diagnosis studied and others, as relevant)
7.5 Retention of Study Records
Copies of protocols, case report forms, correspondence, informed consents and other documents relevant to the study will be archived by the chief investigator and retained for at least 7 years after the completion or discontinuation of the study.
7.6 Changes to the Protocol
The study must be conducted as defined in the present protocol. All changes must be documented by signed protocol amendments or a revised protocol, which will be submitted to the appropriate REC for approval. The chief investigator is responsible for notifying and obtaining approval from the Ethics Committee and the Regulatory Authorities for any changes to the protocol before implementation. National requirements will be followed.
7.7 Publications
The results of the study will be reported and disseminated in peer reviewed scientific journals and conference presentations.
7.8 Study Termination
The study may be terminated at any time for reasons of safety and tolerability as determined by the chief investigator.